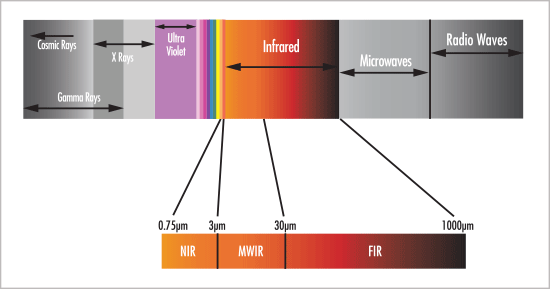
BUCHI NIR Specialist Jason Corbiere met up with BUCHI Market Manager Ryanne Palermo to discuss how benchtop or in-line NIR can address three major areas of concern for QC labs: labor shortages, workplace safety, and slow QC feedback loops. Watch the video discussion here or read the transcript below to learn more. https://youtu.be/xfNYV98EnIg Transcript RP:... Continue Reading →
Addressing labor shortages, workplace safety, and slow QC feedback loops with NIR.